The role of NF-κB in cancer progression, looking at HCC liver cancer.
HCC (Hepatocellular carcinoma) is the third leading cause of cancer deaths worldwide and is the most common form of liver cancer (Thorgeirsson SS and Grisham JW, 2003). Epidemiological studies show that infection with hepatitis B and C virus (HBV and HCV) or contact with genotoxic and cytotoxic chemicals, such as ethanol, are the leading causes of HCC. This results in chronic liver injury and inflammation (Bosch FX, Ribes J et al., 2004). In all of these conditions, hepatocytes are killed and Kupffer cells and inflammatory cells are recruited to the sire and then activated. These cells produce cytokines and bring about compensatory proliferation in the existing hepatocytes. The exact molecular phenomenon behind chronic liver inflammation and its progress to HCC is still to be fully understood (Maeda S, Kamata H et al., 2005).
The connection between NF-κB signalling and cancer was up to recently only circumstantial, as constitutively active NF-κB was present in numerous cancers. Current research has shown that there is a strong genetic and biochemical proof to link NF-κB to cancers. Even so, this cannot be used as a general concept since NF-κB activity primarily depends on the cell type it acts in. Due to this, it can either increase or decrease the extent and development of the disease (Karin M., 2006)
The key role of NF-κB in cancer progression can be attributed to its function of activating anti-apoptotic gene expression (Beg AA and Baltimore D., 1996). When NF-κB gets activated in progressing cancer or pre-neoplastic cells in liver cancer, it allows the survival of malignant cells and restricts their removal by pro-apoptotic surveillance mechanisms. However, in certain cases, activation of NF-κB can bring about a negative effect on the progression of tumor cells as it can have an opposite effect on inflammatory cells (Karin M., 2006). Various regulators play a key role in the inflammation-fibrosis-cancer axis and NF-κB has been implicated as one of them. Also NF-κB has been shown to hold a central role in liver homeostasis, pathophysiology and regulation and progress to HCC. However, to understand the role of NF-κB in HCC, its functions in normal physiology and function should be defined (Sun B and Karin M., 2008).
NF-κB functions in different ways in the liver. When the injury caused to the liver is frequent or chronic, NF-κB has been linked with high levels of pro-inflammatory cytokines, chemokines and matrix mettalloproteinases. This leads to fibrosis and cirrhosis that subsequently elevated the risk of progression to HCC (Elsharkawy AM and Mann DA., 2007). Other studies using knockout mice have shown that NF-κB has a hepatoprotective function and it can safeguard the liver from apoptosis (Ghosh and Karin M., 2002). It also helps to protect hepatocytes and increase their survival potential and liver integrity (Chang L, Kamata H, Solinas G et al., 2006).
NF-κB is a cytoplasmic transcription factor that regulates over 200 genes. It is involved in many cellular and organismal processes including immune and inflammatory responses, developmental processes, cellular growth and apoptosis. It has also been implicated in a large number of disease states such as cancer, arthritis, chronic inflammation, asthma, neurodegeneration and heart diseases (Beyaert R., 2004).
The target genes coded for by NF-κB can be classified into four groups: immunoregulatory and inflammatory genes, anti-apoptotic genes, genes that positively regulate cell proliferation and genes that encode for negative regulators of NF-κB. All of these groups contribute to tumorogenesis (Karin M, Cao Y et al., 2002).
NF-κB as a transcription factor has many diverse functions. At the cellular level, the activation of NF-κB inhibits programmed and elicited cell death. This protects the cell and allows it to survive. It also induces expression of crucial genes that encode for anti-apoptotic proteins such gas Bcl-XL, c-FLIP, A1/Bfl etc. This upholds cell survival (Lin A, Karin M., 2003). It also controls the expression of anti-oxidant genes such as ferritin heavy chain (Pham CG, Bubici C et al., 2004) and superoxide dismustase 2 (SOD2) (Kamata H, Honda S et al., 2005) that add to its prosurvival functions.
The NF-κB family is composed of five members: p65 or Rel-A, Rel-B, c-Rel, nfκb1 (p50/p105), which is a combination of p50 and its precursor p105, nfκb2 (p52/p100), which is a complex between p52 and its precursor 100. The inactive precursor, p100 and p105 are inhibitory proteins that maintain the NF-κB dimers in the cytosol of a resting cell (Pereira SG and Oakley F., 2008). They have been characterised as such due to the presence of an evolutionarily conserved 300-amino acid Rel Homology Domain (RHD) positioned at their N-terminal ends (Hayden MS and Ghosh S., 2004). Rel-A, Rel-B and c-Rel have transcriptional activation domains (TADs) in their C-terminal ends that allow them to activate target gene expression. On the other hand, nfκb1 and nfκb2 do not contain TADs and so they repress transcription unless they are bound to a protein that has a TAD such as the other three NF-κB family members (Hoffmann A, Natoli G and Ghosh G., 2006).
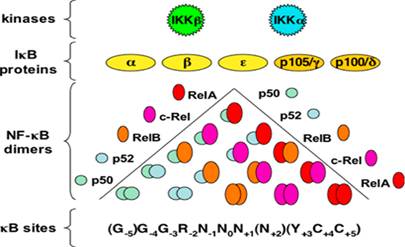
The NF-κB family members exist as either homodimers or heterodimers bound to the family of proteins in an unstimulated cell. The IκB family restricts the translocation of NF-κB to the nucleus, thereby maintaining it in its inactive state. Members of the IκB family have many copies of a sequence called the ankyrin repeat domains that cover the nuclear localisation sequence of the NF-κB members and prevent them from entering into the nucleus (Hayden MS and Ghosh S., 2004).
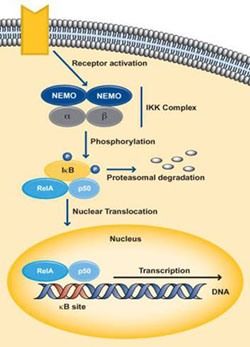
NF-κB is usually present in its latent, inactive form unless stimulated by an environmental response. Through the binding of NF-κB to T-cell receptors, B-cell receptors, TNFR, CD40, BAFFR, LTbR and Toll/IL-1R family, the function of various cytokines, growth factors and effector enzymes get regulated. It can also control the expression of genes and factors outside the immune system (Hayden MS and Ghosh S., 2004). NF-κB activation is initiated by the signal-induced degradation of IκB proteins by the activation of a kinase called the IκB kinase or IKK. The IKK proteins phosphorylated the IκB followed by quick ubiquitin-dependant degradation by the 26S proteosome (Karin M and Ben-neriah Y., 2001). The IKK is made up of IKKα and IKKβ, which are two catalytic subunits as well as IKKγ or NEMO (NF-κB essential modulator), which is a vital non-enzymatic regulatory subunit (Lin A and Karin M., 2003).
When the cell receives any one of a multitude of external stimuli or certain receptors are recognised, NF-κB signaling can occur either by the classical pathway (canonical) or the alternate pathway (non-canonical) (Hayden MS and Ghosh S., 2004) (Karin M., 1999).
The classical pathway gets activated by cytokines such as IL-1 and TNF-α or by bacterial products such as LPS or when a ligand binds to one of the TLR superfamily receptors. Activation of the receptor causes proteins like TRAF to get recruited to the cytoplasmic doman of the receptor. These adaptor proteins cause the activation of the IKK complex, which in the canonical pathway consists of one molecule of IKKα, one molecule IKKβ and two molecules of NEMO. IKKβ phosphorylates IκBα at serine residues 32 and 36 resulting in poly-ubiquitination followed by degradation by the 26S proteosome. This releases NF-κB which is predominantly p50:p65 from its inhibitor IκBα, which then translocares into the nucleus and activates the target genes regulated by the κB sites (Hayden and Ghosh., 2004) (Perkins ND., 2006). Dimers made up of Rel-A, Rel-B, c-Rel and p50 are usually activated by the classical pathway.
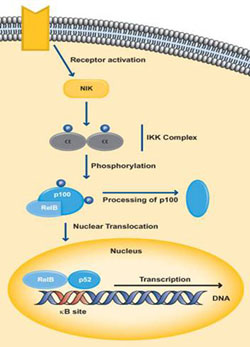
 The non-canonical pathway activates NF-κB complexes of p100/Rel-B and mainly occurs during the development of lymphoid organs responsible for the production of T and B lymphocytes. Very few stimuli can activate NF-κB through this pathway such as B cell activating factor, Lymphotoxin B and CD40. The IKK complex in this pathway is only made up of two subunits of IKKa. Activation by the ligand, stimulates NF-κB inducing kinase or NIK which in turn phosphorylates the IkB site of p100 which results in the processing and release of the p52/Rel-B active heterodimer (Hayden MS, Ghosh., 2004) (Moynagh P., 2005). This active heterodimer translocates into the nucleus and causes target gene expression.
The non-canonical pathway activates NF-κB complexes of p100/Rel-B and mainly occurs during the development of lymphoid organs responsible for the production of T and B lymphocytes. Very few stimuli can activate NF-κB through this pathway such as B cell activating factor, Lymphotoxin B and CD40. The IKK complex in this pathway is only made up of two subunits of IKKa. Activation by the ligand, stimulates NF-κB inducing kinase or NIK which in turn phosphorylates the IkB site of p100 which results in the processing and release of the p52/Rel-B active heterodimer (Hayden MS, Ghosh., 2004) (Moynagh P., 2005). This active heterodimer translocates into the nucleus and causes target gene expression.
 The NF-κB family member looked into in this project is the nfκb1 gene. It codes for a functional protein p50 and its inactive precursor p105 and the gene is located on chromosome 4q24. The p105 subunit covers 3452bp and 25 exons. It also coded for a protein made of 971 amino acids. The p50 subunit occupies the 1-430 amino acids of p105 (Perkins, 2006). The Rel Homology domain of p50 consists of N-terminal domain (NTD), dimerization domain (DimD) and nuclear localisation sequence (NLS) that controls DNA binding, dimerisation and nuclear localisation respectively. The stop signal for the proteosome is provided by the Glycine-rich region (GRR) located adjacent to RHD. This aids in the stabilisation of the newly generated p50 and prevents its proteolytic breakdown (Lin L, DeMartino GN and Greene W., 1998) (Moorthy AK, Savino OV et al., 2006).
The NF-κB family member looked into in this project is the nfκb1 gene. It codes for a functional protein p50 and its inactive precursor p105 and the gene is located on chromosome 4q24. The p105 subunit covers 3452bp and 25 exons. It also coded for a protein made of 971 amino acids. The p50 subunit occupies the 1-430 amino acids of p105 (Perkins, 2006). The Rel Homology domain of p50 consists of N-terminal domain (NTD), dimerization domain (DimD) and nuclear localisation sequence (NLS) that controls DNA binding, dimerisation and nuclear localisation respectively. The stop signal for the proteosome is provided by the Glycine-rich region (GRR) located adjacent to RHD. This aids in the stabilisation of the newly generated p50 and prevents its proteolytic breakdown (Lin L, DeMartino GN and Greene W., 1998) (Moorthy AK, Savino OV et al., 2006).
The way in which p50 is generated is still contentious. Even so, it is understandable that co-translation and post-translational processes mediate the production and formation of p50 homodimers (Pereira SG, Oakley F., 2006).
The co-translation of p50 is done by GRR. The death domains (DD) present in the C-terminal end allows the interaction between p105 and IKKs. Activated IKKs will phophorylate p105 at serine residues which causes ubiquitination and proteolytic degradation of p105 by the 26S proteosome and releases p50 (Lin L, DeMartino GN and Greene W., 1998) (Moorthy et al., 2006). This implies that p105 and IkBγ can bind and maintain the dimers in an inactive state in the cytoplasm (Perkins ND, 2006). Numerous controversies regarding whether the nfκb1 gene is a transcriptional repressor or activators exists. There is enough proof to advocate both, so it has been decided that p50 can carry out both functions. Their ability to function as a transcriptional repressor can be because they lack a transactivational domain (Ghosh S, May MJ and Kopp EB., 1998) and they have the ability to recruit HDAC1 (histone deacetylase-1) to promoters when p50 is a homodimer. To behave as an activator of transcription, nfκb1 homodimers need external stimuli.
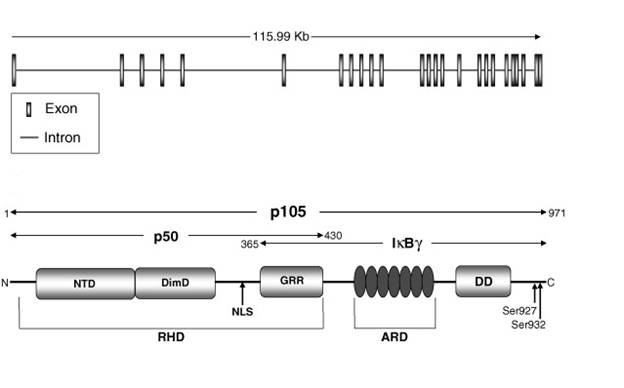
The p105 precursor contains the N-terminal domain (NTD), dimerization doman (DimD), glycine-rich ankyrin repeats (GRR), ankyrin repeat domain (ARD) and the death domain (DD) (Perkins N, 2006). The death domain has been found to facilitate the interaction between p105 and the IKKs. At serines 927 and 932, which allow ubiquitination and proteolytic breakdown of p105 by the 26S proteosome and release of p50 (Lin et al., 1998) (Moorthy et al., 2006). The ankyrin repeats present also inhibit activation of NF-κB, as they possess sequences homologous to the ARD in the IκBa (Pereira SG and Oakley F., 2006)
Another important fact about p50 is that its homodimers can bind to naked DNA as well as nucleosomal DNA which allows it to regulate transcriptionally repressed gene expression along with active chromatin (Angelov, F Lenouvel, F Hans, CW Muller et al., 2004).
For this project, more important than the mechanism by which the active form of nfκ1 gene is generated is the inflammatory and fibrogenic response it initiates to chronic injury. Also, since p50 homodimers have been implicated in repressing transcription, a lot of concentration in this project is towards understanding this mechanism and the effect it has on cytokines, growth factors and immune cells.
Recently, there has been substantial evidence that each sub-unit of NF-κB has distinct roles and they influence the growth of tumors and production of inflammatory mediators. Homodimers of p50 have the ability to act as active repressors of inflammation. The absence of the transactivation domain requires p50 to form heterodimers with p65 or c-Rel to be transcriptionally active (Ghosh S, May MJ and Kopp EB., 1998). On the other hand p50 homodimers do preserve their ability to bind to NF-κB sites and are implicated to be repressors of transcription (May MJ, Ghosh S., 1997). This can be clearly seen when macrophages were stimulated with LPS or Toll-like receptor agonists. A complex is formed between p50 homodimers and transcriptional co-activator CREB binding protein which afterwards binds to a cis-element in the IL-10 promoter region and sets off transcription. This results in an amplified expression of anti-inflammatory cytokines (Cao S, Zhang X et al., 2006).
As for it being a transcriptional repressor, studies have shown that in the absence of the nfκb1 gene in mouse models causes aberrant production of TNF-α. This showed us that p50 homodimers and the HDAC1 form a complex that function as an active repressor of TNF-α activity (Peirera SG and Oakley F., 2006).
Keeping this in mind, this study has focused on the nfκb1 gene and its relevance is investigated using a well-established mouse model of chemically induced hepatocellular carcinoma by administration of diethylnitrosoamine (DEN), which is a liver carcinogen. DEN, when injected into the mice gets metabolised to form an alkylating agent that causes DNA damage as well as mutations along with hepatocyte death. (Farber JL and Gerson RJ., 1984) Also, previous data has demonstrated that in hepatocytes NF-κB plays an important role in the continued existence of hepatocytes. (Shin M, Lufen C et al., 2003)
The way NF-κB functions in cancer can be attributed to which particular subunit is most active in the cell type. In the liver the most common and active form of NF-κB is the heterodimer of RelA and p50. Studies have already confirmed that nfκb1 gene plays a hepatoprotective role in Carbon tetrachloride (CCl4) -liver injury models. Studies conducted by Oakley et al, on CCl4-injured livers shows that the nfκb1 gene was an important regulator of tissue inflammation in response to injury and infection. Using this model, it was also evident that the absence of the nfκb1 gene is associated with intense hepatic inflammation and progress to a harsh fibrogenic response. It was concluded in this study that the nfκb1 gene played a protective role in chronic liver injury and the mechanism behind this was attributed to TNF-α expression by p50 homodimer-HDAC1 complex (Oakley F, Mann J et al., 2005).
This concept was extended into my project and the protective role of nfκb1 gene was studied in a DEN-hepatocellular carcinoma mouse model. Using acute as well as chronic DEN-injury models, the differences in inflammatory cells, growth factors, cell cycle regulators and cytokine expression was compared between the nfκb1-/- and nfκb+/+ mice.
BIBLIOGRAPHY
Bosch FX, Ribes J, Díaz M, Cléries R.,. (2004). Primary liver cancer: worldwide incidence and trends, Gastroenterology, 127: 5-16
Maeda, S., Kamata, H., Luo, J-L, Leffert and H., Karin, M., (2005). IKK Couples Hepatocyte Cell Death to Cytokine-driven Compensatory Proliferation that Promotes Chemical Hepatocarcinogenesis. Cell. 121:977-990
Michael Karin. (2006) Nuclear factor-kB in cancer development and progression, Nature, 441:431-436
Beg, AA and Baltimore , D. (1996) An essential role of NF-κB in preventing TNF-α induced cell death, Science 274, 782-784
B Sun and Michael Karin. (2008) nfκb signaling, liver disease and hepatoprotective agents. Oncogene. 27: 6228-6244
Elsharkawy AM, Mann DA (2007). Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 46: 590-597
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K et al., (2006) The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP (L) turnover. Cell 124:601-613.
Ghosh and Karin M (2002). Missing pieces in the NF-kappa B puzzle.Cell 109: 81-96.
Karin M, Cao Y, Greten FR, Li ZW, (2002) NF-κB in cancer: from innocent bystander to major culprit, Nature reviews cancer,. 2: 301-310
Beyaert R (2004). Nuclear Factor-kappaB: Regulation and Role in Disease. Kluwer Academic Publishes, Dordrecht, The Netherlands. 426 pages
Cao, S., Zhang, X., Edwards, J. P., & Mosser, D. M. (2006). NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem., 281, 26041–26050
Farber JL, Gerson RJ. (1984) Mechanisms of cell injury with hepatotoxic chemicals, Pharmacol Rev, 36: 71-75
Angelov, F. Lenouvel, F. Hans, C.W. Muller, P. Bouvet and J. Bednar et al.,. (2002) The histone octamer is invisible when NF-kappaB binds to the nucleosome. J. Biol. Chem. 279: 42374–42382
Moynagh P. N. (2005) The Nfκb pathway. J Cell Sci. 118, 4389-4392
Perkins ND. (2006) Integrating cell-signalling pathways with NF-κB and IKK function, Nat. Rev. 8: 49–62
Pereira SG Oakley F. (2008) Nuclear factor-kappaB1: Regulation and function. The international journal of biochemistry & cell biology.40: 1425-30.
Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K et al. (2004) Ferritin heavy chain unregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119: 529-542
May MJ, Ghosh S., (1997) Rel/NF-kappa B and I kappa B proteins: An overview, Seminars in cancer biology. 8: 63-73
Lin A, Karin M (2003). NF-kappaB in cancer:a marked target. Semin Cancer Biol. 13:107-114
L. Lin, G.N. DeMartino and W. Greene, (1998) Co-translational biogenesis of NF-κB p50 by the 26S proteasome, Cell 92: 819–828
Karin M. (1999) How Nfκb is activated: the role of the IkB kinase (IKK) complex. Oncogene. 18, 6867-6874
Karin M, Cao Y, Greten FR, Li ZW, (2002) NF-κB in cancer: from innocent bystander to major culprit, Nature reviews cancer,. 2: 301-310
Karin M, ben-neriah Y. (2001) Proliferation, cell cycle and apoptosis in cancer. Nature; 411:342-8
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005). Reactive oxygen species promote TNFalpha induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120:649-661
Hoffmann A., Natoli G., Ghosh G. (2006) Transcriptional regulation via the Nfκb signaling module. Oncogene. 25, 6706-6716.
Hayden and Ghosh.(2004) Signaling to nfκb, Genes and development, 18:2195-2224
Ghosh S, May MJ, Kopp EB. (1998) NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses, Annual review of Immunology, 16:225-260
Fiona Oakley , Jelena Mann, Sarah Nailard, David E Smart, NArendra Mungalsingh, Christothea Constandinou, Shakir Ali, Susan J Wilson, Harry Millward-Sadler, John P Iredale and Derek A Mann. (2005) Nuclear factor-kB1 (p50) limits the inflammatory and fibrogenic response to chronic injury, American Journal of pathology, 166: 695-708
A.K. Moorthy, O.V. Savinova, J. Ho, Y. Wang, D. Vu and G. Gourisankar. (2006) The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner, EMBO J., 25: 1945–1956